11 Inside Dielectrics
Inside Dielectrics
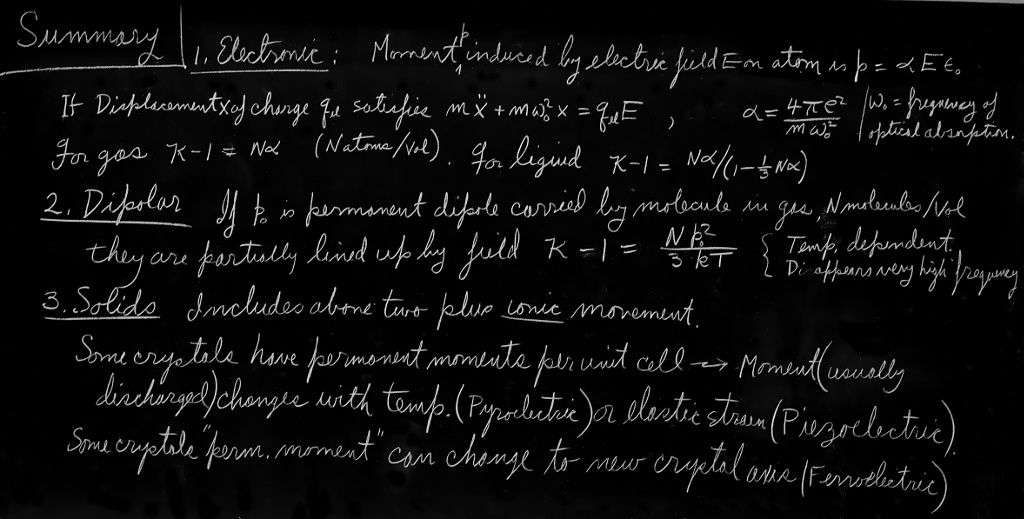
| Review: | Chapter 31, Vol. I, The Origin of the Refractive Index |
| Chapter 40, Vol. I, The Principles of Statistical Mechanics |
11–1Molecular dipoles
In this chapter we are going to discuss why it is that materials are dielectric. We said in the last chapter that we could understand the properties of electrical systems with dielectrics once we appreciated that when an electric field is applied to a dielectric it induces a dipole moment in the atoms. Specifically, if the electric field $E$ induces an average dipole moment per unit volume $P$, then $\kappa$, the dielectric constant, is given by \begin{equation} \label{Eq:II:11:1} \kappa-1=\frac{P}{\epsO E}. \end{equation}
We have already discussed how this equation is applied; now we have to discuss the mechanism by which polarization arises when there is an electric field inside a material. We begin with the simplest possible example—the polarization of gases. But even gases already have complications: there are two types. The molecules of some gases, like oxygen, which has a symmetric pair of atoms in each molecule, have no inherent dipole moment. But the molecules of others, like water vapor (which has a nonsymmetric arrangement of hydrogen and oxygen atoms) carry a permanent electric dipole moment. As we pointed out in Chapter 6, there is in the water vapor molecule an average plus charge on the hydrogen atoms and a negative charge on the oxygen. Since the center of gravity of the negative charge and the center of gravity of the positive charge do not coincide, the total charge distribution of the molecule has a dipole moment. Such a molecule is called a polar molecule. In oxygen, because of the symmetry of the molecule, the centers of gravity of the positive and negative charges are the same, so it is a nonpolar molecule. It does, however, become a dipole when placed in an electric field. The forms of the two types of molecules are sketched in Fig. 11–1.
11–2Electronic polarization
We will first discuss the polarization of nonpolar molecules. We can start with the simplest case of a monatomic gas (for instance, helium). When an atom of such a gas is in an electric field, the electrons are pulled one way by the field while the nucleus is pulled the other way, as shown in Fig. 10–4. Although the atoms are very stiff with respect to the electrical forces we can apply experimentally, there is a slight net displacement of the centers of charge, and a dipole moment is induced. For small fields, the amount of displacement, and so also the dipole moment, is proportional to the electric field. The displacement of the electron distribution which produces this kind of induced dipole moment is called electronic polarization.
We have already discussed the influence of an electric field on an atom in Chapter 31 of Vol. I, when we were dealing with the theory of the index of refraction. If you think about it for a moment, you will see that what we must do now is exactly the same as we did then. But now we need worry only about fields that do not vary with time, while the index of refraction depended on time-varying fields.
In Chapter 31 of Vol. I we supposed that when an atom is placed in an oscillating electric field the center of charge of the electrons obeys the equation \begin{equation} \label{Eq:II:11:2} m\,\frac{d^2x}{dt^2}+m\omega_0^2x=q_eE. \end{equation} The first term is the electron mass times its acceleration and the second is a restoring force, while the right-hand side is the force from the outside electric field. If the electric field varies with the frequency $\omega$, Eq. (11.2) has the solution \begin{equation} \label{Eq:II:11:3} x=\frac{q_eE}{m(\omega_0^2-\omega^2)}, \end{equation} which has a resonance at $\omega=\omega_0$. When we previously found this solution, we interpreted it as saying that $\omega_0$ was the frequency at which light (in the optical region or in the ultraviolet, depending on the atom) was absorbed. For our purposes, however, we are interested only in the case of constant fields, i.e., for $\omega=0$, so we can disregard the acceleration term in (11.2), and we find that the displacement is \begin{equation} \label{Eq:II:11:4} x=\frac{q_eE}{m\omega_0^2}. \end{equation}
From this we see that the dipole moment $p$ of a single atom is \begin{equation} \label{Eq:II:11:5} p=q_ex=\frac{q_e^2E}{m\omega_0^2}. \end{equation} In this theory the dipole moment $p$ is indeed proportional to the electric field.
People usually write \begin{equation} \label{Eq:II:11:6} \FLPp=\alpha\epsO\FLPE. \end{equation} (Again the $\epsO$ is put in for historical reasons.) The constant $\alpha$ is called the polarizability of the atom, and has the dimensions $L^3$. It is a measure of how easy it is to induce a moment in an atom with an electric field. Comparing (11.5) and (11.6), our simple theory says that \begin{equation} \label{Eq:II:11:7} \alpha=\frac{q_e^2}{\epsO m\omega_0^2}=\frac{4\pi e^2}{m\omega_0^2}. \end{equation}
If there are $N$ atoms in a unit volume, the polarization $P$—the dipole moment per unit volume—is given by \begin{equation} \label{Eq:II:11:8} \FLPP=N\FLPp=N\alpha\epsO\FLPE. \end{equation}
Putting (11.1) and (11.8) together, we get \begin{equation} \label{Eq:II:11:9} \kappa-1=\frac{P}{\epsO E}=N\alpha \end{equation} or, using (11.7), \begin{equation} \label{Eq:II:11:10} \kappa-1=\frac{4\pi Ne^2}{m\omega_0^2}. \end{equation}
From Eq. (11.10) we would predict that the dielectric constant $\kappa$ of different gases should depend on the density of the gas and on the frequency $\omega_0$ of its optical absorption.
Our formula is, of course, only a very rough approximation, because in Eq. (11.2) we have taken a model which ignores the complications of quantum mechanics. For example, we have assumed that an atom has only one resonant frequency, when it really has many. To calculate properly the polarizability $\alpha$ of atoms we must use the complete quantum-mechanical theory, but the classical ideas above give us a reasonable estimate.
Let’s see if we can get the right order of magnitude for the dielectric constant of some substance. Suppose we try hydrogen. We have once estimated (Chapter 38, Vol. I) that the energy needed to ionize the hydrogen atom should be approximately \begin{equation} \label{Eq:II:11:11} E\approx\frac{1}{2}\,\frac{me^4}{\hbar^2}. \end{equation} For an estimate of the natural frequency $\omega_0$, we can set this energy equal to $\hbar\omega_0$—the energy of an atomic oscillator whose natural frequency is $\omega_0$. We get \begin{equation*} \omega_0\approx\frac{1}{2}\,\frac{me^4}{\hbar^3}. \end{equation*} If we now use this value of $\omega_0$ in Eq. (11.7), we find for the electronic polarizability \begin{equation} \label{Eq:II:11:12} \alpha\approx16\pi\biggl[\frac{\hbar^2}{me^2}\biggr]^3. \end{equation} The quantity $(\hbar^2/me^2)$ is the radius of the ground-state orbit of a Bohr atom (see Chapter 38, Vol. I) and equals $0.529$ angstroms. In a gas at standard pressure and temperature ($1$ atmosphere, $0^\circ$C) there are $2.69\times10^{19}$ atoms/cm$^3$, so Eq. (11.9) gives us \begin{equation} \label{Eq:II:11:13} \kappa=1+(2.69\times10^{19})16\pi(0.529\times10^{-8})^3=1.00020. \end{equation} \begin{align} \kappa&=1+(2.69\times10^{19})16\pi(0.529\times10^{-8})^3\notag\\[1ex] \label{Eq:II:11:13} &=1.00020. \end{align}
The dielectric constant for hydrogen gas is measured to be \begin{equation*} \kappa_{\text{exp}}=1.00026. \end{equation*} We see that our theory is about right. We should not expect any better, because the measurements were, of course, made with normal hydrogen gas, which has diatomic molecules, not single atoms. We should not be surprised if the polarization of the atoms in a molecule is not quite the same as that of the separate atoms. The molecular effect, however, is not really that large. An exact quantum-mechanical calculation of $\alpha$ for hydrogen atoms gives a result about $12\%$ higher than (11.12) (the $16\pi$ is changed to $18\pi$), and therefore predicts a dielectric constant somewhat closer to the observed one. In any case, it is clear that our model of a dielectric is fairly good.
Another check on our theory is to try Eq. (11.7) on atoms which have a higher frequency of excitation. For instance, it takes about $24.6$ electron volts to pull the electron off helium, compared with the $13.6$ electron volts required to ionize hydrogen. We would, therefore, expect that the absorption frequency $\omega_0$ for helium would be about twice as big as for hydrogen and that $\alpha$ would be one-quarter as large. So, from (11.13) we expect that \begin{equation*} \kappa_{\text{helium}}\approx1.000050. \end{equation*} Experimentally, \begin{equation*} \kappa_{\text{helium}}=1.000068, \end{equation*} so you see that our rough estimates are coming out on the right track. So we have understood the dielectric constant of nonpolar gas, but only qualitatively, because we have not yet used a correct atomic theory of the motions of the atomic electrons.
11–3Polar molecules; orientation polarization
Next we will consider a molecule which carries a permanent dipole moment $p_0$—such as a water molecule. With no electric field, the individual dipoles point in random directions, so the net moment per unit volume is zero. But when an electric field is applied, two things happen: First, there is an extra dipole moment induced because of the forces on the electrons; this part gives just the same kind of electronic polarizability we found for a nonpolar molecule. For very accurate work, this effect should, of course, be included, but we will neglect it for the moment. (It can always be added in at the end.) Second, the electric field tends to line up the individual dipoles to produce a net moment per unit volume. If all the dipoles in a gas were to line up, there would be a very large polarization, but that does not happen. At ordinary temperatures and electric fields the collisions of the molecules in their thermal motion keep them from lining up very much. But there is some net alignment, and so some polarization (see Fig. 11–2). The polarization that does occur can be computed by the methods of statistical mechanics we described in Chapter 40 of Vol. I.
To use this method we need to know the energy of a dipole in an electric field. Consider a dipole of moment $\FLPp_0$ in an electric field, as shown in Fig. 11–3. The energy of the positive charge is $q\phi(1)$, and the energy of the negative charge is $-q\phi(2)$. Thus the energy of the dipole is \begin{equation} U=q\phi(1)-q\phi(2)=q\FLPd\cdot\FLPgrad{\phi},\notag \end{equation} or \begin{equation} \label{Eq:II:11:14} U=-\FLPp_0\cdot\FLPE=-p_0E\cos\theta, \end{equation} where $\theta$ is the angle between $\FLPp_0$ and $\FLPE$. As we would expect, the energy is lower when the dipoles are lined up with the field.
We now find out how much lining up occurs by using the methods of statistical mechanics. We found in Chapter 40 of Vol. I that in a state of thermal equilibrium, the relative number of molecules with the potential energy $U$ is proportional to \begin{equation} \label{Eq:II:11:15} e^{-U/kT}, \end{equation} where $U(x,y,z)$ is the potential energy as a function of position. The same arguments would say that using Eq. (11.14) for the potential energy as a function of angle, the number of molecules at $\theta$ per unit solid angle is proportional to $e^{-U/kT}$.
Letting $n(\theta)$ be the number of molecules per unit solid angle at $\theta$, we have \begin{equation} \label{Eq:II:11:16} n(\theta)=n_0e^{+p_0E\cos\theta/kT}. \end{equation} For normal temperatures and fields, the exponent is small, so we can approximate by expanding the exponential: \begin{equation} \label{Eq:II:11:17} n(\theta)=n_0\biggl(1+\frac{p_0E\cos\theta}{kT}\biggr). \end{equation}
We can find $n_0$ if we integrate (11.17) over all angles; the result should be just $N$, the total number of molecules per unit volume. The average value of $\cos\theta$ over all angles is zero, so the integral is just $n_0$ times the total solid angle $4\pi$. We get \begin{equation} \label{Eq:II:11:18} n_0=\frac{N}{4\pi}. \end{equation}
We see from (11.17) that there will be more molecules oriented along the field ($\cos\theta=1$) than against the field ($\cos\theta=-1$). So in any small volume containing many molecules there will be a net dipole moment per unit volume—that is, a polarization $P$. To calculate $P$, we want the vector sum of all the molecular moments in a unit volume. Since we know that the result is going to be in the direction of $\FLPE$, we will just sum the components in that direction (the components at right angles to $\FLPE$ will sum to zero): \begin{equation*} P=\underset{\substack{\text{unit}\\\text{volume}}}{\sum} p_0\cos\theta_i. \end{equation*}
We can evaluate the sum by integrating over the angular distribution. The solid angle at $\theta$ is $2\pi\sin\theta\,d\theta$, so \begin{equation} \label{Eq:II:11:19} P=\int_0^\pi n(\theta)p_0\cos\theta\,2\pi\sin\theta\,d\theta. \end{equation} Substituting for $n(\theta)$ from (11.17), we have \begin{equation*} P=-\frac{N}{2}\int_1^{-1} \biggl(1+\frac{p_0E}{kT}\cos\theta\biggr) p_0\cos\theta\,d(\cos\theta), \end{equation*} which is easily integrated to give \begin{equation} \label{Eq:II:11:20} P=\frac{Np_0^2E}{3kT}. \end{equation} The polarization is proportional to the field $E$, so there will be normal dielectric behavior. Also, as we expect, the polarization depends inversely on the temperature, because at higher temperatures there is more disalignment by collisions. This $1/T$ dependence is called Curie’s law. The permanent moment $p_0$ appears squared for the following reason: In a given electric field, the aligning force depends upon $p_0$, and the mean moment that is produced by the lining up is again proportional to $p_0$. The average induced moment is proportional to $p_0^2$.
We should now try to see how well Eq. (11.20) agrees with experiment. Let’s look at the case of steam. Since we don’t know what $p_0$ is, we cannot compute $P$ directly, but Eq. (11.20) does predict that $\kappa-1$ should vary inversely as the temperature, and this we should check.
From (11.20) we get \begin{equation} \label{Eq:II:11:21} \kappa-1=\frac{P}{\epsO E}=\frac{Np_0^2}{3\epsO kT}, \end{equation} so $\kappa-1$ should vary in direct proportion to the density $N$, and inversely as the absolute temperature. The dielectric constant has been measured at several different pressures and temperatures, chosen such that the number of molecules in a unit volume remained fixed.1 [Notice that if the measurements had all been taken at constant pressure, the number of molecules per unit volume would decrease linearly with increasing temperature and $\kappa-1$ would vary as $T^{-2}$ instead of as $T^{-1}$.] In Fig. 11–4 we plot the experimental observations for $\kappa-1$ as a function of $1/T$. The dependence predicted by (11.21) is followed quite well.
There is another characteristic of the dielectric constant of polar molecules—its variation with the frequency of the applied field. Due to the moment of inertia of the molecules, it takes a certain amount of time for the heavy molecules to turn toward the direction of the field. So if we apply frequencies in the high microwave region or above, the polar contribution to the dielectric constant begins to fall away because the molecules cannot follow. In contrast to this, the electronic polarizability still remains the same up to optical frequencies, because of the smaller inertia in the electrons.
11–4Electric fields in cavities of a dielectric
We now turn to an interesting but complicated question—the problem of the dielectric constant in dense materials. Suppose that we take liquid helium or liquid argon or some other nonpolar material. We still expect electronic polarization. But in a dense material, $\FLPP$ can be large, so the field on an individual atom will be influenced by the polarization of the atoms in its close neighborhood. The question is, what electric field acts on the individual atom?
Imagine that the liquid is put between the plates of a condenser. If the plates are charged they will produce an electric field in the liquid. But there are also charges in the individual atoms, and the total field $\FLPE$ is the sum of both of these effects. This true electric field varies very, very rapidly from point to point in the liquid. It is very high inside the atoms—particularly right next to the nucleus—and relatively small between the atoms. The potential difference between the plates is the line integral of this total field. If we ignore all the fine-grained variations, we can think of an average electric field $E$, which is just $V/d$. (This is the field we were using in the last chapter.) We should think of this field as the average over a space containing many atoms.
Now you might think that an “average” atom in an “average” location would feel this average field. But it is not that simple, as we can show by considering what happens if we imagine different-shaped holes in a dielectric. For instance, suppose that we cut a slot in a polarized dielectric, with the slot oriented parallel to the field, as shown in part (a) of Fig. 11–5. Since we know that $\FLPcurl{\FLPE}=\FLPzero$, the line integral of $\FLPE$ around the curve, $\Gamma$, which goes as shown in (b) of the figure, should be zero. The field inside the slot must give a contribution which just cancels the part from the field outside. Therefore the field $E_0$ actually found in the center of a long thin slot is equal to $E$, the average electric field found in the dielectric.
Now consider another slot whose large sides are perpendicular to $E$, as shown in part (c) of Fig. 11–5. In this case, the field $E_0$ in the slot is not the same as $E$ because polarization charges appear on the surfaces. If we apply Gauss’ law to a surface $S$ drawn as in (d) of the figure, we find that the field $E_0$ in the slot is given by \begin{equation} \label{Eq:II:11:22} E_0=E+\frac{P}{\epsO}, \end{equation} where $E$ is again the electric field in the dielectric. (The Gaussian surface contains the surface polarization charge $\sigma_{\text{pol}}=P$.) We mentioned in Chapter 10 that $\epsO E+P$ is often called $D$, so $\epsO E_0=D_0$ is equal to $D$ in the dielectric.
Earlier in the history of physics, when it was supposed to be very important to define every quantity by direct experiment, people were delighted to discover that they could define what they meant by $E$ and $D$ in a dielectric without having to crawl around between the atoms. The average field $\FLPE$ is numerically equal to the field $\FLPE_0$ that would be measured in a slot cut parallel to the field. And the field $\FLPD$ could be measured by finding $E_0$ in a slot cut normal to the field. But nobody ever measures them that way anyway, so it was just one of those philosophical things.
For most liquids which are not too complicated in structure, we could expect that an atom finds itself, on the average, surrounded by the other atoms in what would be a good approximation to a spherical hole. And so we should ask: “What would be the field in a spherical hole?” We can find out by noticing that if we imagine carving out a spherical hole in a uniformly polarized material, we are just removing a sphere of polarized material. (We must imagine that the polarization is “frozen in” before we cut out the hole.) By superposition, however, the fields inside the dielectric, before the sphere was removed, is the sum of the fields from all charges outside the spherical volume plus the fields from the charges within the polarized sphere. That is, if we call $E$ the field in the uniform dielectric, we can write \begin{equation} \label{Eq:II:11:23} E=E_{\text{hole}}+E_{\text{plug}}, \end{equation} where $E_{\text{hole}}$ is the field in the hole and $E_{\text{plug}}$ is the field inside a sphere which is uniformly polarized (see Fig. 11–6). The fields due to a uniformly polarized sphere are shown in Fig. 11–7. The electric field inside the sphere is uniform, and its value is \begin{equation} \label{Eq:II:11:24} E_{\text{plug}}=-\frac{P}{3\epsO}. \end{equation} Using (11.23), we get \begin{equation} \label{Eq:II:11:25} E_{\text{hole}}=E+\frac{P}{3\epsO}. \end{equation} The field in a spherical cavity is greater than the average field by the amount $P/3\epsO$. (The spherical hole gives a field $1/3$ of the way between a slot parallel to the field and a slot perpendicular to the field.)
11–5The dielectric constant of liquids; the Clausius-Mossotti equation
In a liquid we expect that the field which will polarize an individual atom is more like $E_{\text{hole}}$ than just $E$. If we use the $E_{\text{hole}}$ of (11.25) for the polarizing field in Eq. (11.6), then Eq. (11.8) becomes \begin{equation} \label{Eq:II:11:26} P=N\alpha\epsO\biggl(E+\frac{P}{3\epsO}\biggr), \end{equation} or \begin{equation} \label{Eq:II:11:27} P=\frac{N\alpha}{1-(N\alpha/3)}\,\epsO E. \end{equation} Remembering that $\kappa-1$ is just $P/\epsO E$, we have \begin{equation} \label{Eq:II:11:28} \kappa-1=\frac{N\alpha}{1-(N\alpha/3)}, \end{equation} which gives us the dielectric constant of a liquid in terms of $\alpha$, the atomic polarizability. This is called the Clausius-Mossotti equation.
Whenever $N\alpha$ is very small, as it is for a gas (because the density $N$ is small), then the term $N\alpha/3$ can be neglected compared with $1$, and we get our old result, Eq. (11.9), that \begin{equation} \label{Eq:II:11:29} \kappa-1=N\alpha. \end{equation}
Let’s compare Eq. (11.28) with some experimental results. It is first necessary to look at gases for which, using the measurement of $\kappa$, we can find $\alpha$ from Eq. (11.29). For instance, for carbon disulfide at zero degrees centigrade the dielectric constant is $1.0029$, so $N\alpha$ is $0.0029$. Now the density of the gas is easily worked out and the density of the liquid can be found in handbooks. At $20^\circ$C, the density of liquid CS$_2$ is $381$ times higher than the density of the gas at $0^\circ$C. This means that $N$ is $381$ times higher in the liquid than it is in the gas, so that—if we make the approximation that the basic atomic polarizability of the carbon disulfide doesn’t change when it is condensed into a liquid—$N\alpha$ in the liquid is equal to $381$ times $0.0029$, or $1.11$. Notice that the $N\alpha/3$ term amounts to almost $0.4$, so it is quite significant. With these numbers we predict a dielectric constant of $2.76$, which agrees reasonably well with the observed value of $2.64$.
In Table 11–1 we give some experimental data on various materials (taken from the Handbook of Chemistry and Physics), together with the dielectric constants calculated from Eq. (11.28) in the way just described. The agreement between observation and theory is even better for argon and oxygen than for CS$_2$—and not so good for carbon tetrachloride. On the whole, the results show that Eq. (11.28) works very well.
| Gas | Liquid | |||||||
| Substance | κ(exp) | Nα | Density | Density | Ratio1 | Nα | κ (predict) | κ (exp) |
| CS2 | $1.0029\phantom{00}$ | $0.0029\phantom{00}$ | $0.00339$ | $1.293$ | $381$ | $1.11\phantom{0}$ | $2.76\phantom{0}$ | $2.64\phantom{0}$ |
| O2 | $1.000523$ | $0.000523$ | $0.00143$ | $1.19\phantom{0}$ | $832$ | $0.435$ | $1.509$ | $1.507$ |
| CCl4 | $1.0030\phantom{00}$ | $0.0030\phantom{00}$ | $0.00489$ | $1.59\phantom{0}$ | $325$ | $0.977$ | $2.45\phantom{0}$ | $2.24\phantom{0}$ |
| Ar | $1.000545$ | $0.000545$ | $0.00178$ | $1.44\phantom{0}$ | $810$ | $0.441$ | $1.517$ | $1.54\phantom{0}$ |
| 1Ratio = density of liquid/density of gas. | ||||||||
Our derivation of Eq. (11.28) is valid only for electronic polarization in liquids. It is not right for a polar molecule like H$_2$O. If we go through the same calculations for water, we get $13.2$ for $N\alpha$, which means that the dielectric constant for the liquid is negative, while the observed value of $\kappa$ is $80$. The problem has to do with the correct treatment of the permanent dipoles, and Onsager has pointed out the right way to go. We do not have the time to treat the case now, but if you are interested it is discussed in Kittel’s book, Introduction to Solid State Physics.
11–6Solid dielectrics
Now we turn to the solids. The first interesting fact about solids is that there can be a permanent polarization built in—which exists even without applying an electric field. An example occurs with a material like wax, which contains long molecules having a permanent dipole moment. If you melt some wax and put a strong electric field on it when it is a liquid, so that the dipole moments get partly lined up, they will stay that way when the liquid freezes. The solid material will have a permanent polarization which remains when the field is removed. Such a solid is called an electret.
An electret has permanent polarization charges on its surface. It is the electrical analog of a magnet. It is not as useful, though, because free charges from the air are attracted to its surfaces, eventually cancelling the polarization charges. The electret is “discharged” and there are no visible external fields.
A permanent internal polarization $P$ is also found occurring naturally in some crystalline substances. In such crystals, each unit cell of the lattice has an identical permanent dipole moment, as drawn in Fig. 11–8. All the dipoles point in the same direction, even with no applied electric field. Many complicated crystals have, in fact, such a polarization; we do not normally notice it because the external fields are discharged, just as for the electrets.
If these internal dipole moments of a crystal are changed, however, external fields appear because there is not time for stray charges to gather and cancel the polarization charges. If the dielectric is in a condenser, free charges will be induced on the electrodes. For example, the moments can change when a dielectric is heated, because of thermal expansion. The effect is called pyroelectricity. Similarly, if we change the stresses in a crystal—for instance, if we bend it—again the moment may change a little bit, and a small electrical effect, called piezoelectricity, can be detected.
For crystals that do not have a permanent moment, one can work out a theory of the dielectric constant that involves the electronic polarizability of the atoms. It goes much the same as for liquids. Some crystals also have rotatable dipoles inside, and the rotation of these dipoles will also contribute to $\kappa$. In ionic crystals such as NaCl there is also ionic polarizability. The crystal consists of a checkerboard of positive and negative ions, and in an electric field the positive ions are pulled one way and the negatives the other; there is a net relative motion of the plus and minus charges, and so a volume polarization. We could estimate the magnitude of the ionic polarizability from our knowledge of the stiffness of salt crystals, but we will not go into that subject here.
11–7Ferroelectricity; BaTiO$_{\boldsymbol{3}}$
We want to describe now one special class of crystals which have, just by accident almost, a built-in permanent moment. The situation is so marginal that if we increase the temperature a little bit they lose the permanent moment completely. On the other hand, if they are nearly cubic crystals, so that their moments can be turned in different directions, we can detect a large change in the moment when an applied electric field is changed. All the moments flip over and we get a large effect. Substances which have this kind of permanent moment are called ferroelectric, after the corresponding ferromagnetic effects which were first discovered in iron.
We would like to explain how ferroelectricity works by describing a particular example of a ferroelectric material. There are several ways in which the ferroelectric property can originate; but we will take up only one mysterious case—that of barium titanate, BaTiO$_3$. This material has a crystal lattice whose basic cell is sketched in Fig. 11–9. It turns out that above a certain temperature, specifically $118^\circ$C, barium titanate is an ordinary dielectric with an enormous dielectric constant. Below this temperature, however, it suddenly takes on a permanent moment.
In working out the polarization of solid material, we must first find what are the local fields in each unit cell. We must include the fields from the polarization itself, just as we did for the case of a liquid. But a crystal is not a homogeneous liquid, so we cannot use for the local field what we would get in a spherical hole. If you work it out for a crystal, you find that the factor $1/3$ in Eq. (11.24) becomes slightly different, but not far from $1/3$. (For a simple cubic crystal, it is just $1/3$.) We will, therefore, assume for our preliminary discussion that the factor is $1/3$ for BaTiO$_3$.
Now when we wrote Eq. (11.28) you may have wondered what would happen if $N\alpha$ became greater than $3$. It appears as though $\kappa$ would become negative. But that surely cannot be right. Let’s see what should happen if we were gradually to increase $\alpha$ in a particular crystal. As $\alpha$ gets larger, the polarization gets bigger, making a bigger local field. But a bigger local field will polarize each atom more, raising the local fields still more. If the “give” of the atoms is enough, the process keeps going; there is a kind of feedback that causes the polarization to increase without limit—assuming that the polarization of each atom increases in proportion to the field. The “runaway” condition occurs when $N\alpha=3$. The polarization does not become infinite, of course, because the proportionality between the induced moment and the electric field breaks down at high fields, so that our formulas are no longer correct. What happens is that the lattice gets “locked in” with a high, self-generated, internal polarization.
In the case of BaTiO$_3$, there is, in addition to an electronic polarization, also a rather large ionic polarization, presumed to be due to titanium ions which can move a little within the cubic lattice. The lattice resists large motions, so after the titanium has gone a little way, it jams up and stops. But the crystal cell is then left with a permanent dipole moment.
In most crystals, this is really the situation for all temperatures that can be reached. The very interesting thing about barium titanate is that there is such a delicate condition that if $N\alpha$ is decreased just a little bit it comes unstuck. Since $N$ decreases with increasing temperature—because of thermal expansion—we can vary $N\alpha$ by varying the temperature. Below the critical temperature it is just barely stuck, so it is easy—by applying an external field—to shift the polarization and have it lock in a different direction.
Let’s see if we can analyze what happens in more detail. We call $T_c$ the critical temperature at which $N\alpha$ is exactly $3$. As the temperature increases, $N$ goes down a little bit because of the expansion of the lattice. Since the expansion is small, we can say that near the critical temperature \begin{equation} \label{Eq:II:11:30} N\alpha=3-\beta(T-T_c), \end{equation} where $\beta$ is a small constant, of the same order of magnitude as the thermal expansion coefficient, or about $10^{-5}$ to $10^{-6}$ per degree C. Now if we substitute this relation into Eq. (11.28), we get that \begin{equation*} \kappa-1=\frac{3-\beta(T-T_c)}{\beta(T-T_c)/3}. \end{equation*} Since we have assumed that $\beta(T-T_c)$ is small compared with one, we can approximate this formula by \begin{equation} \label{Eq:II:11:31} \kappa-1=\frac{9}{\beta(T-T_c)}. \end{equation}
This relation is right, of course, only for $T>T_c$. We see that just above the critical temperature $\kappa$ is enormous. Because $N\alpha$ is so close to $3$, there is a tremendous magnification effect, and the dielectric constant can easily be as high as $50{,}000$ to $100{,}000$. It is also very sensitive to temperature. For increases in temperature, the dielectric constant goes down inversely as the temperature, but, unlike the case of a dipolar gas, for which $\kappa-1$ goes inversely as the absolute temperature, for ferroelectrics it varies inversely as the difference between the absolute temperature and the critical temperature (this law is called the Curie-Weiss law).
When we lower the temperature to the critical temperature, what happens? If we imagine a lattice of unit cells like that in Fig. 11–9, we see that it is possible to pick out chains of ions along vertical lines. One of them consists of alternating oxygen and titanium ions. There are other lines made up of either barium or oxygen ions, but the spacing along these lines is greater. We make a simple model to imitate this situation by imagining, as shown in Fig. 11–10(a), a series of chains of ions. Along what we call the main chain, the separation of the ions is $a$, which is half the lattice constant; the lateral distance between identical chains is $2a$. There are less-dense chains in between which we will ignore for the moment. To make the analysis a little easier, we will also suppose that all the ions on the main chain are identical. (It is not a serious simplification because all the important effects will still appear. This is one of the tricks of theoretical physics. One does a different problem because it is easier to figure out the first time—then when one understands how the thing works, it is time to put in all the complications.)
Now let’s try to find out what would happen with our model. We suppose that the dipole moment of each atom is $p$ and we wish to calculate the field at one of the atoms of the chain. We must find the sum of the fields from all the other atoms. We will first calculate the field from the dipoles in only one vertical chain; we will talk about the other chains later. The field at the distance $r$ from a dipole in a direction along its axis is given by \begin{equation} \label{Eq:II:11:32} E=\frac{1}{4\pi\epsO}\,\frac{2p}{r^3}. \end{equation} At any given atom, the dipoles at equal distances above and below it give fields in the same direction, so for the whole chain we get \begin{equation} \label{Eq:II:11:33} E_{\text{chain}}=\frac{p}{4\pi\epsO}\,\frac{2}{a^3}\cdot \biggl(2+\frac{2}{8}+\frac{2}{27}+\frac{2}{64}+\dotsb\biggr)= \frac{p}{\epsO}\,\frac{0.383}{a^3}. \end{equation} \begin{align} E_{\text{chain}}&=\frac{p}{4\pi\epsO}\,\frac{2}{a^3}\cdot \biggl(2+\frac{2}{8}+\frac{2}{27}+\frac{2}{64}+\dotsb\biggr)\notag\\[1.5ex] \label{Eq:II:11:33} &=\;\,\frac{p}{\epsO}\,\frac{0.383}{a^3}. \end{align} It is not too hard to show that if our model were like a completely cubic crystal—that is, if the next identical lines were only the distance $a$ away—the number $0.383$ would be changed to $1/3$. In other words, if the next lines were at the distance $a$ they would contribute only $-0.050$ unit to our sum. However, the next main chain we are considering is at the distance $2a$ and, as you remember from Chapter 7, the field from a periodic structure dies off exponentially with distance. Therefore these lines contribute much less than $-0.050$ and we can just ignore all the other chains.
It is necessary now to find out what polarizability $\alpha$ is needed to make the runaway process work. Suppose that the induced moment $p$ of each atom of the chain is proportional to the field on it, as in Eq. (11.6). We get the polarizing field on the atom from $E_{\text{chain}}$ using Eq. (11.32). So we have the two equations \begin{equation*} p=\alpha\epsO E_{\text{chain}} \end{equation*} and \begin{equation*} E_{\text{chain}}=\frac{0.383}{a^3}\,\frac{p}{\epsO}. \end{equation*} There are two solutions: $E_{\text{chain}}$ and $p$ both zero, or \begin{equation*} \alpha=\frac{a^3}{0.383}, \end{equation*} with $E_{\text{chain}}$ and $p$ both finite. Thus if $\alpha$ is as large as $a^3/0.383$, a permanent polarization sustained by its own field will set in. This critical equality must be reached for barium titanate at just the temperature $T_c$. (Notice that if $\alpha$ were larger than the critical value for small fields, it would decrease at larger fields and at equilibrium the same equality we have found would hold.)
For BaTiO$_3$, the spacing $a$ is $2\times10^{-8}$ cm, so we must expect that $\alpha=21.8\times10^{-24}$ cm$^3$. We can compare this with the known polarizabilities of the individual atoms. For oxygen, $\alpha=30.2\times10^{-24}$ cm$^3$; we’re on the right track! But for titanium, $\alpha=2.4\times10^{-24}$ cm$^3$; rather small. To use our model we should probably take the average. (We could work out the chain again for alternating atoms, but the result would be about the same.) So $\alpha(\text{average})=16.3\times10^{-24}$ cm$^3$, which is not high enough to give a permanent polarization.
But wait a moment! We have so far only added up the electronic polarizabilities. There is also some ionic polarization due to the motion of the titanium ion. All we need is an ionic polarizability of $9.2\times10^{-24}$ cm$^3$. (A more precise computation using alternating atoms shows that actually $11.9\times10^{-24}$ cm$^3$ is needed.) To understand the properties of BaTiO$_3$, we have to assume that such an ionic polarizability exists.
Why the titanium ion in barium titanate should have that much ionic polarizability is not known. Furthermore, why, at a lower temperature, it polarizes along the cube diagonal and the face diagonal equally well is not clear. If we figure out the actual size of the spheres in Fig. 11–9, and ask whether the titanium is a little bit loose in the box formed by its neighboring oxygen atoms—which is what you would hope, so that it could be easily shifted—you find quite the contrary. It fits very tightly. The barium atoms are slightly loose, but if you let them be the ones that move, it doesn’t work out. So you see that the subject is really not one-hundred percent clear; there are still mysteries we would like to understand.
Returning to our simple model of Fig. 11–10(a), we see that the field from one chain would tend to polarize the neighboring chain in the opposite direction, which means that although each chain would be locked, there would be no net permanent moment per unit volume! (Although there would be no external electric effects, there are still certain thermodynamic effects one could observe.) Such systems exist, and are called antiferroelectric. So what we have explained is really an antiferroelectric. Barium titanate, however, is really like the arrangement in Fig. 11–10(b). The oxygen-titanium chains are all polarized in the same direction because there are intermediate chains of atoms in between. Although the atoms in these chains are not very polarizable, or very dense, they will be somewhat polarized, in the direction antiparallel to the oxygen-titanium chains. The small fields produced at the next oxygen-titanium chain will get it started parallel to the first. So BaTiO$_3$ is really ferroelectric, and it is because of the atoms in between. You may be wondering: “But what about the direct effect between the two O-Ti chains?” Remember, though, the direct effect dies off exponentially with the separation; the effect of the chain of strong dipoles at $2a$ can be less than the effect of a chain of weak ones at the distance $a$.
This completes our rather detailed report on our present understanding of the dielectric constants of gases, of liquids, and of solids.
- Sänger, Steiger, and Gächter, Helvetica Physica Acta 5, 200 (1932). ↩