32 Refractive Index of Dense Materials
Refractive Index of Dense Materials
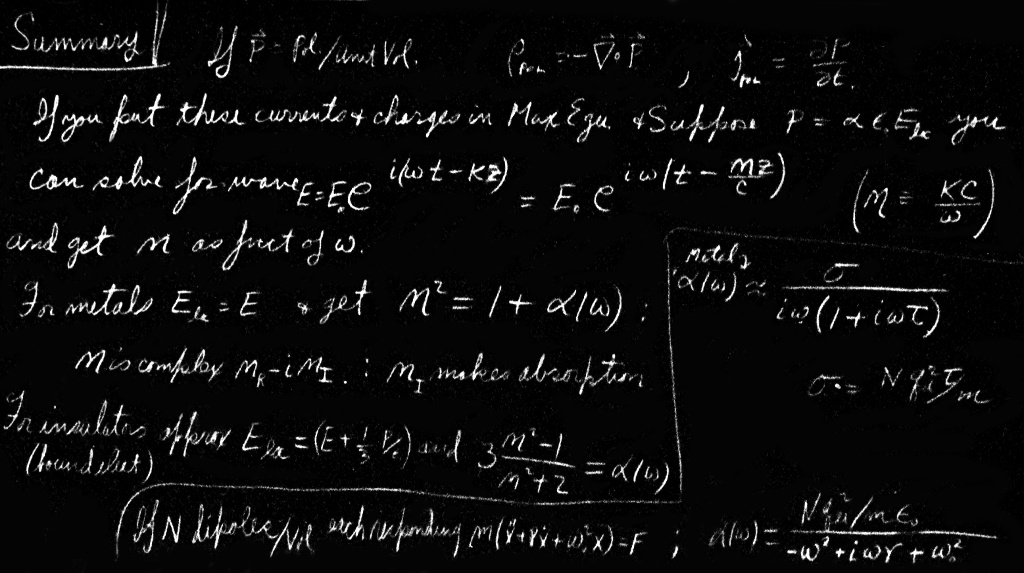
| Review: | See Table 32–1. |
32–1Polarization of matter
We want now to discuss the phenomenon of the refraction of light—and also, therefore, the absorption of light—by dense materials. In Chapter 31 of Volume I we discussed the theory of the index of refraction, but because of our limited mathematical abilities at that time, we had to restrict ourselves to finding the index only for materials of low density, like gases. The physical principles that produced the index were, however, made clear. The electric field of the light wave polarizes the molecules of the gas, producing oscillating dipole moments. The acceleration of the oscillating charges radiates new waves of the field. This new field, interfering with the old field, produces a changed field which is equivalent to a phase shift of the original wave. Because this phase shift is proportional to the thickness of the material, the effect is equivalent to having a different phase velocity in the material. When we looked at the subject before, we neglected the complications that arise from such effects as the new wave changing the fields at the oscillating dipoles. We assumed that the forces on the charges in the atoms came just from the incoming wave, whereas, in fact, their oscillations are driven not only by the incoming wave but also by the radiated waves of all the other atoms. It would have been difficult for us at that time to include this effect, so we studied only the rarefied gas, where such effects are not important.
Now, however, we will find that it is very easy to treat the problem by the use of differential equations. This method obscures the physical origin of the index (as coming from the re-radiated waves interfering with the original waves), but it makes the theory for dense materials much simpler. This chapter will bring together a large number of pieces from our earlier work. We’ve taken up practically everything we will need, so there are relatively few really new ideas to be introduced. Since you may need to refresh your memory about what we are going to need, we give in Table 32–1 a list of the equations we are going to use, together with a reference to the place where each can be found. In most instances, we will not take the time to give the physical arguments again, but will just use the equations.
| Subject | Reference | Equation |
| Damped oscillations | Vol. I, Chap. 23 | $m(\ddot{x}+\gamma\dot{x}+\omega_0^2x)=F$ |
| Index of gases | Vol. I, Chap. 31 | $n=1+\dfrac{1}{2}\,\dfrac{Nq_e^2}{\epsO m(\omega_0^2-\omega^2)}$ |
| $n=n'-in''$ | ||
| Mobility | Vol. I, Chap. 41 | $m\ddot{x}+\mu\dot{x}=F$ |
| Electrical conductivity | Vol. I, Chap. 43 | $\mu=\dfrac{\tau}{m};\,\sigma=\dfrac{Nq_e^2\tau}{m}$ |
| Polarizability | Vol. II, Chap. 10 | $\rho_{\text{pol}}=-\FLPdiv{\FLPP}$ |
| Inside dielectrics | Vol. II, Chap. 11 | $\FLPE_{\text{local}}=\FLPE+\dfrac{1}{3\epsO}\,\FLPP$ |
We begin by recalling the machinery of the index of refraction for a gas. We suppose that there are $N$ particles per unit volume and that each particle behaves as a harmonic oscillator. We use a model of an atom or molecule in which the electron is bound with a force proportional to its displacement (as though the electron were held in place by a spring). We emphasized that this was not a legitimate classical model of an atom, but we will show later that the correct quantum mechanical theory gives results equivalent to this model (in simple cases). In our earlier treatment, we did not include the possibility of a damping force in the atomic oscillators, but we will do so now. Such a force corresponds to a resistance to the motion, that is, to a force proportional to the velocity of the electron. Then the equation of motion is \begin{equation} \label{Eq:II:32:1} F=q_eE=m(\ddot{x}+\gamma\dot{x}+\omega_0^2x), \end{equation} where $x$ is the displacement parallel to the direction of $\FLPE$. (We are assuming an isotropic oscillator whose restoring force is the same in all directions. Also, we are taking, for the moment, a linearly polarized wave, so that $\FLPE$ doesn’t change direction.) If the electric field acting on the atom varies sinusoidally with time, we write \begin{equation} \label{Eq:II:32:2} E=E_0e^{i\omega t}. \end{equation} The displacement will then oscillate with the same frequency, and we can let \begin{equation*} x=x_0e^{i\omega t}. \end{equation*} Substituting $\dot{x}=i\omega x$ and $\ddot{x}=-\omega^2x$, we can solve for $x$ in terms of $E$: \begin{equation} \label{Eq:II:32:3} x=\frac{q_e/m}{-\omega^2+i\gamma\omega+\omega_0^2}\,E. \end{equation} Knowing the displacement, we can calculate the acceleration $\ddot{x}$ and find the radiated wave responsible for the index. This was the way we computed the index in Chapter 31 of Volume I.
Now, however, we want to take a different approach. The induced dipole moment $p$ of an atom is $q_ex$ or, using Eq. (32.3), \begin{equation} \label{Eq:II:32:4} \FLPp=\frac{q_e^2/m}{-\omega^2+i\gamma\omega+\omega_0^2}\,\FLPE. \end{equation} Since $\FLPp$ is proportional to $\FLPE$, we write \begin{equation} \label{Eq:II:32:5} \FLPp=\epsO\alpha(\omega)\FLPE, \end{equation} where $\alpha$ is called the atomic polarizability.1 With this definition, we have \begin{equation} \label{Eq:II:32:6} \alpha=\frac{q_e^2/m\epsO}{-\omega^2+i\gamma\omega+\omega_0^2}. \end{equation}
The quantum mechanical solution for the motions of electrons in atoms gives a similar answer except with the following modifications. The atoms have several natural frequencies, each frequency with its own dissipation constant $\gamma$. Also the effective “strength” of each mode is different, which we can represent by multiplying the polarizability for each frequency by a strength factor $f$, which is a number we expect to be of the order of $1$. Representing the three parameters $\omega_0$, $\gamma$, and $f$ by $\omega_{0k}$, $\gamma_k$, and $f_k$ for each mode of oscillation, and summing over the various modes, we modify Eq. (32.6) to read \begin{equation} \label{Eq:II:32:7} \alpha(\omega)=\frac{q_e^2}{\epsO m} \sum_k\frac{f_k}{-\omega^2+i\gamma_k\omega+\omega_{0k}^2}. \end{equation}
If $N$ is the number of atoms per unit volume in the material, the polarization $P$ is just $Np=\epsO N\alpha E$, and is proportional to $E$: \begin{equation} \label{Eq:II:32:8} \FLPP=\epsO N\alpha(\omega)\FLPE. \end{equation} In other words, when there is a sinusoidal electric field acting in a material, there is an induced dipole moment per unit volume which is proportional to the electric field—with a proportionality constant $\alpha$ that, we emphasize, depends upon the frequency. At very high frequencies, $\alpha$ is small; there is not much response. However, at low frequencies there can be a strong response. Also, the proportionality constant is a complex number, which means that the polarization does not exactly follow the electric field, but may be shifted in phase to some extent. At any rate, there is a polarization per unit volume whose magnitude is proportional to the strength of the electric field.
32–2Maxwell’s equations in a dielectric
The existence of polarization in matter means that there are polarization charges and currents inside of the material, and these must be put into the complete Maxwell equations in order to find the fields. We are going to solve Maxwell’s equations this time in a situation in which the charges and currents are not zero, as in a vacuum, but are given implicitly by the polarization vector. Our first step is to find explicitly the charge density $\rho$ and current density $\FLPj$, averaged over a small volume of the same size we had in mind when we defined $\FLPP$. Then the $\rho$ and $\FLPj$ we need can be obtained from the polarization.
We have seen in Chapter 10 that when the polarization $\FLPP$ varies from place to place, there is a charge density given by \begin{equation} \label{Eq:II:32:9} \rho_{\text{pol}}=-\FLPdiv{\FLPP}. \end{equation} At that time, we were dealing with static fields, but the same formula is valid also for time-varying fields. However, when $\FLPP$ varies with time, there are charges in motion, so there is also a polarization current. Each of the oscillating charges contributes a current equal to its charge $q_e$, times its velocity $v$. With $N$ such charges per unit volume, the current density $\FLPj$ is \begin{equation*} \FLPj=Nq_e\FLPv. \end{equation*} Since we know that $v=dx/dt$, then $j=Nq_e(dx/dt)$, which is just $dP/dt$. Therefore the current density from the varying polarization is \begin{equation} \label{Eq:II:32:10} \FLPj_{\text{pol}}=\ddp{\FLPP}{t}. \end{equation}
Our problem is now direct and simple. We write Maxwell’s equations with the charge density and current density expressed in terms of $\FLPP$, using Eqs. (32.9) and (32.10). (We assume that there are no other currents and charges in the material.) We then relate $\FLPP$ to $\FLPE$ with Eq. (32.8), and we solve the equation for $\FLPE$ and $\FLPB$—looking for the wave solutions.
Before we do this, we would like to make an historical note. Maxwell originally wrote his equations in a form which was different from the one we have been using. Because the equations were written in this different form for many years—and are still written that way by many people—we will explain the difference. In the early days, the mechanism of the dielectric constant was not fully and clearly appreciated. The nature of atoms was not understood, nor that there was a polarization of the material. So people did not appreciate that there was a contribution to the charge density $\rho$ from $\FLPdiv{\FLPP}$. They thought only in terms of charges that were not bound to atoms (such as the charges that flow in wires or are rubbed off surfaces).
Today, we prefer to let $\rho$ represent the total charge density, including the part from the bound atomic charges. If we call that part $\rho_{\text{pol}}$, we can write \begin{equation*} \rho=\rho_{\text{pol}}+\rho_{\text{other}}, \end{equation*} where $\rho_{\text{other}}$ is the charge density considered by Maxwell and refers to the charges not bound to individual atoms. We would then write \begin{equation*} \FLPdiv{\FLPE}=\frac{\rho_{\text{pol}}+\rho_{\text{other}}}{\epsO}. \end{equation*} Substituting $\rho_{\text{pol}}$ from Eq. (32.9), \begin{equation} \FLPdiv{\FLPE}=\frac{\rho_{\text{other}}}{\epsO}- \frac{1}{\epsO}\,\FLPdiv{\FLPP}\notag \end{equation} or \begin{equation} \label{Eq:II:32:11} \FLPdiv{(\epsO\FLPE+\FLPP)}=\rho_{\text{other}}. \end{equation}
The current density in the Maxwell equations for $\FLPcurl{\FLPB}$ also has, in general, contributions from bound atomic currents. We can therefore write \begin{equation*} \FLPj=\FLPj_{\text{pol}}+\FLPj_{\text{other}}, \end{equation*} and the Maxwell equation becomes \begin{equation} \label{Eq:II:32:12} c^2\FLPcurl{\FLPB}=\frac{\FLPj_{\text{other}}}{\epsO}+ \frac{\FLPj_{\text{pol}}}{\epsO}+\ddp{\FLPE}{t}. \end{equation} Using Eq. (32.10), we get \begin{equation} \label{Eq:II:32:13} \epsO c^2\FLPcurl{\FLPB}=\FLPj_{\text{other}}+ \ddp{}{t}(\epsO\FLPE+\FLPP). \end{equation}
Now you can see that if we were to define a new vector $\FLPD$ by \begin{equation} \label{Eq:II:32:14} \FLPD=\epsO\FLPE+\FLPP, \end{equation} the two field equations would become \begin{equation} \label{Eq:II:32:15} \FLPdiv{\FLPD}=\rho_{\text{other}} \end{equation} and \begin{equation} \label{Eq:II:32:16} \epsO c^2\FLPcurl{\FLPB}=\FLPj_{\text{other}}+\ddp{\FLPD}{t}. \end{equation} These are actually the forms that Maxwell used for dielectrics. His two remaining equations were \begin{equation*} \FLPcurl{\FLPE}=-\ddp{\FLPB}{t}, \end{equation*} and \begin{equation*} \FLPdiv{\FLPB}=0, \end{equation*} which are the same as we have been using.
Maxwell and the other early workers also had a problem with magnetic materials (which we will take up soon). Because they did not know about the circulating currents responsible for atomic magnetism, they used a current density that was missing still another part. Instead of Eq. (32.16), they actually wrote \begin{equation} \label{Eq:II:32:17} \FLPcurl{\FLPH}=\FLPj'+\ddp{\FLPD}{t}, \end{equation} where $\FLPH$ differs from $\epsO c^2\FLPB$ because it includes the effects of atomic currents. (Then $\FLPj'$ represents what is left of the currents.) So Maxwell had four field vectors—$\FLPE$, $\FLPD$, $\FLPB$, and $\FLPH$—the $\FLPD$ and $\FLPH$ were hidden ways of not paying attention to what was going on inside the material. You will find the equations written this way in many places.
To solve the equations, it is necessary to relate $\FLPD$ and $\FLPH$ to the other fields, and people used to write \begin{equation} \label{Eq:II:32:18} \FLPD=\epsilon\FLPE\quad\text{and}\quad \FLPB=\mu\FLPH. \end{equation} However, these relations are only approximately true for some materials and even then only if the fields are not changing rapidly with time. (For sinusoidally varying fields one often can write the equations this way by making $\epsilon$ and $\mu$ complex functions of the frequency, but not for an arbitrary time variation of the fields.) So there used to be all kinds of cheating in solving the equations. We think the right way is to keep the equations in terms of the fundamental quantities as we now understand them—and that’s how we have done it.
32–3Waves in a dielectric
We want now to find out what kind of electromagnetic waves can exist in a dielectric material in which there are no extra charges other than those bound in atoms. So we take $\rho=-\FLPdiv{\FLPP}$ and $\FLPj=\ddpl{\FLPP}{t}$. Maxwell’s equations then become \begin{equation} \label{Eq:II:32:19} \begin{array}{llll} (\text{a}) & \FLPdiv{\FLPE}=-\dfrac{\FLPdiv{\FLPP}}{\epsO}\quad & (\text{b}) & c^2\FLPcurl{\FLPB}=\displaystyle\ddp{}{t}\biggl(\dfrac{\FLPP}{\epsO}+ \FLPE\biggr)\\ \\ (\text{c}) & \FLPcurl{\FLPE}=-\displaystyle\ddp{\FLPB}{t} & (\text{d}) & \FLPdiv{\FLPB}=0 \end{array} \end{equation} \begin{equation} \begin{aligned} &(\text{a})\;\;\FLPdiv{\FLPE}=-\dfrac{\FLPdiv{\FLPP}}{\epsO}\\[1ex] &(\text{b})\;\;c^2\FLPcurl{\FLPB}=\displaystyle\ddp{}{t}\biggl(\dfrac{\FLPP}{\epsO}+\FLPE\biggr)\\[1ex] &(\text{c})\;\;\FLPcurl{\FLPE}=-\displaystyle\ddp{\FLPB}{t}\\[1.5ex] &(\text{d})\;\;\FLPdiv{\FLPB}=0 \end{aligned} \label{Eq:II:32:19} \end{equation}
We can solve these equations as we have done before. We start by taking the curl of Eq. (32.19c): \begin{equation*} \FLPcurl{(\FLPcurl{\FLPE})}=-\ddp{}{t}\,\FLPcurl{\FLPB}. \end{equation*} Next, we make use of the vector identity \begin{equation*} \FLPcurl{(\FLPcurl{\FLPE})}=\FLPgrad{(\FLPdiv{\FLPE})}-\nabla^2\FLPE, \end{equation*} and also substitute for $\FLPcurl{\FLPB}$, using Eq. (32.19b); we get \begin{equation*} \FLPgrad{(\FLPdiv{\FLPE})}-\nabla^2\FLPE= -\frac{1}{\epsO c^2}\,\frac{\partial^2\FLPP}{\partial t^2}- \frac{1}{c^2}\,\frac{\partial^2\FLPE}{\partial t^2}. \end{equation*} Using Eq. (32.19a) for $\FLPdiv{\FLPE}$, we get \begin{equation} \label{Eq:II:32:20} \nabla^2\FLPE-\frac{1}{c^2}\,\frac{\partial^2\FLPE}{\partial t^2}= -\frac{1}{\epsO}\,\FLPgrad{(\FLPdiv{\FLPP})}+ \frac{1}{\epsO c^2}\,\frac{\partial^2\FLPP}{\partial t^2}. \end{equation} \begin{align} \label{Eq:II:32:20} \nabla^2\FLPE&-\frac{1}{c^2}\,\frac{\partial^2\FLPE}{\partial t^2}=\\[1ex] &-\frac{1}{\epsO}\,\FLPgrad{(\FLPdiv{\FLPP})}+ \frac{1}{\epsO c^2}\,\frac{\partial^2\FLPP}{\partial t^2}.\notag \end{align} So instead of the wave equation, we now get that the d’Alembertian of $\FLPE$ is equal to two terms involving the polarization $\FLPP$.
Since $\FLPP$ depends on $\FLPE$, however, Eq. (32.20) can still have wave solutions. We will now limit ourselves to isotropic dielectrics, so that $\FLPP$ is always in the same direction as $\FLPE$. Let’s try to find a solution for a wave going in the $z$-direction. Then, the electric field might vary as $e^{i(\omega t-kz)}$. We will also suppose that the wave is polarized in the $x$-direction—that the electric field has only an $x$-component. We write \begin{equation} \label{Eq:II:32:21} E_x=E_0e^{i(\omega t-kz)}. \end{equation}
You know that any function of $(z-vt)$ represents a wave that travels with the speed $v$. The exponent of Eq. (32.21) can be written as \begin{equation*} -ik\biggl(z-\frac{\omega}{k}\,t\biggr), \end{equation*} so, Eq. (32.21) represents a wave with the phase velocity \begin{equation*} v_{\text{ph}}=\omega/k. \end{equation*} The index of refraction $n$ is defined (see Chapter 31, Vol. I) by letting \begin{equation*} v_{\text{ph}}=\frac{c}{n}. \end{equation*} Thus Eq. (32.21) becomes \begin{equation*} E_x=E_0e^{i\omega(t-nz/c)}. \end{equation*} So we can find $n$ by finding what value of $k$ is required if Eq. (32.21) is to satisfy the proper field equations, and then using \begin{equation} \label{Eq:II:32:22} n=\frac{kc}{\omega}. \end{equation} In an isotropic material, there will be only an $x$-component of the polarization; then $\FLPP$ has no variation with the $x$-coordinate, so $\FLPdiv{\FLPP}=0$, and we get rid of the first term on the right-hand side of Eq. (32.20). Also, since we are assuming a linear dielectric, $P_x$ will vary as $e^{i\omega t}$, and $\partial^2P_x/\partial t^2=-\omega^2P_x$. The Laplacian in Eq. (32.20) becomes simply $\partial^2E_x/\partial z^2=-k^2E_x$, so we get \begin{equation} \label{Eq:II:32:23} -k^2E_x+\frac{\omega^2}{c^2}\,E_x=-\frac{\omega^2}{\epsO c^2}\,P_x. \end{equation}
Now let us assume for the moment that since $\FLPE$ is varying sinusoidally, we can set $\FLPP$ proportional to $\FLPE$, as in Eq. (32.8). (We’ll come back to discuss this assumption later.) We write \begin{equation*} P_x=\epsO N\alpha E_x. \end{equation*} Then $E_x$ drops out of Eq. (32.23), and we find \begin{equation} \label{Eq:II:32:24} k^2=\frac{\omega^2}{c^2}\,(1+N\alpha). \end{equation} We have found that a wave like Eq. (32.21), with the wave number $k$ given by Eq. (32.24), will satisfy the field equations. Using Eq. (32.22), the index $n$ is given by \begin{equation} \label{Eq:II:32:25} n^2=1+N\alpha. \end{equation}
Let’s compare this formula with what we obtained in our theory of the index of a gas (Chapter 31, Vol. I). There, we got Eq. (31.19), which is \begin{equation} \label{Eq:II:32:26} n=1+\frac{1}{2}\,\frac{Nq_e^2}{m\epsO}\,\frac{1}{-\omega^2+\omega_0^2}. \end{equation} Taking $\alpha$ from Eq. (32.6), Eq. (32.25) would give us \begin{equation} \label{Eq:II:32:27} n^2=1+\frac{Nq_e^2}{m\epsO}\,\frac{1}{-\omega^2+i\gamma\omega+\omega_0^2}. \end{equation} First, we have the new term in $i\gamma\omega$, because we are including the dissipation of the oscillators. Second, the left-hand side is $n$ instead of $n^2$, and there is an extra factor of $1/2$. But notice that if $N$ is small enough so that $n$ is close to one (as it is for a gas), then Eq. (32.27) says that $n^2$ is one plus a small number: $n^2=1+\epsilon$. We can then write $n=\sqrt{1+\epsilon}\approx1+\epsilon/2$, and the two expressions are equivalent. Thus our new method gives for a gas the same result we found earlier.
Now you might think that Eq. (32.27) should give the index of refraction for dense materials also. It needs to be modified, however, for several reasons. First, the derivation of this equation assumes that the polarizing field on each atom is the field $E_x$. That assumption is not right, however, because in dense materials there is also the field produced by other atoms in the vicinity, which may be comparable to $E_x$. We considered a similar problem when we studied the static fields in dielectrics. (See Chapter 11.) You will remember that we estimated the field at a single atom by imagining that it sat in a spherical hole in the surrounding dielectric. The field in such a hole—which we called the local field—is increased over the average field $E$ by the amount $P/3\epsO$. (Remember, however, that this result is only strictly true in isotropic materials—including the special case of a cubic crystal.)
The same arguments will hold for the electric field in a wave, so long as the wavelength of the wave is much longer than the spacing between atoms. Limiting ourselves to such cases, we write \begin{equation} \label{Eq:II:32:28} E_{\text{local}}=E+\frac{P}{3\epsO}. \end{equation} This local field is the one that should be used for $E$ in Eq. (32.3); that is, Eq. (32.8) should be rewritten: \begin{equation} \label{Eq:II:32:29} P=\epsO N\alpha E_{\text{local}}. \end{equation} Using $E_{\text{local}}$ from Eq. (32.28), we find \begin{equation} P=\epsO N\alpha\biggl(E+\frac{P}{3\epsO}\biggr)\notag \end{equation} or \begin{equation} \label{Eq:II:32:30} P=\frac{N\alpha}{1-(N\alpha/3)}\,\epsO E. \end{equation} In other words, for dense materials $P$ is still proportional to $E$ (for sinusoidal fields). However, the constant of proportionality is not $\epsO N\alpha$, as we wrote below Eq. (32.23), but should be $\epsO N\alpha/[1-(N\alpha/3)]$. So we should correct Eq. (32.25) to read \begin{equation} \label{Eq:II:32:31} n^2=1+\frac{N\alpha}{1-(N\alpha/3)}. \end{equation} It will be more convenient if we rewrite this equation as \begin{equation} \label{Eq:II:32:32} 3\,\frac{n^2-1}{n^2+2}=N\alpha, \end{equation} which is algebraically equivalent. This is known as the Clausius-Mossotti equation.
There is another complication in dense materials. Because neighboring atoms are so close, there are strong interactions between them. The internal modes of oscillation are, therefore, modified. The natural frequencies of the atomic oscillations are spread out by the interactions, and they are usually quite heavily damped—the resistance coefficient becomes quite large. So the $\omega_0$’s and $\gamma$’s of the solid will be quite different from those of the free atoms. With these reservations, we can still represent $\alpha$, at least approximately, by Eq. (32.7). We have then that \begin{equation} \label{Eq:II:32:33} 3\,\frac{n^2-1}{n^2+2}=\frac{Nq_e^2}{m\epsO} \sum_k\frac{f_k}{-\omega^2+i\gamma_k\omega+\omega_{0k}^2}. \end{equation}
One final complication. If the dense material is a mixture of several components, each will contribute to the polarization. The total $\alpha$ will be the sum of the contributions from each component of the mixture [except for the inaccuracy of the local field approximation, Eq. (32.28), in ordered crystals—effects we discussed when analyzing ferroelectrics]. Writing $N_j$ as the number of atoms of each component per unit volume, we should replace Eq. (32.32) by \begin{equation} \label{Eq:II:32:34} 3\biggl(\frac{n^2-1}{n^2+2}\biggr)= \sum_jN_j\alpha_j, \end{equation} where each $\alpha_j$ will be given by an expression like Eq. (32.7). Equation (32.34) completes our theory of the index of refraction. The quantity $3(n^2-1)/(n^2+2)$ is given by some complex function of frequency, which is the mean atomic polarizability $\alpha(\omega)$. The precise evaluation of $\alpha(\omega)$ (that is, finding $f_k$, $\gamma_k$ and $\omega_{0k}$) in dense substances is a difficult problem of quantum mechanics. It has been done from first principles only for a few especially simple substances.
32–4The complex index of refraction
We want to look now at the consequences of our result, Eq. (32.33). First, we notice that $\alpha$ is complex, so the index $n$ is going to be a complex number. What does that mean? Let’s say that we write $n$ as the sum of a real and an imaginary part: \begin{equation} \label{Eq:II:32:35} n=n_R-in_I, \end{equation} where $n_R$ and $n_I$ are real functions of $\omega$. We write $in_I$ with a minus sign, so that $n_I$ will be a positive quantity in all ordinary optical materials. (In ordinary inactive materials—that are not, like lasers, light sources themselves—$\gamma$ is a positive number, and that makes the imaginary part of $n$ negative.) Our plane wave of Eq. (32.21) is written in terms of $n$ as \begin{equation*} E_x=E_0e^{i\omega(t-nz/c)}. \end{equation*} Writing $n$ as in Eq. (32.35), we would have \begin{equation} \label{Eq:II:32:36} E_x=E_0e^{-\omega n_Iz/c}e^{i\omega(t-n_Rz/c)}. \end{equation} The term $e^{i\omega(t-n_Rz/c)}$ represents a wave travelling with the speed $c/n_R$, so $n_R$ represents what we normally think of as the index of refraction. But the amplitude of this wave is \begin{equation*} E_0e^{-\omega n_Iz/c}, \end{equation*} which decreases exponentially with $z$. A graph of the strength of the electric field at some instant as a function of $z$ is shown in Fig. 32–1, for $n_I\approx n_R/2\pi$. The imaginary part of the index represents the attenuation of the wave due to the energy losses in the atomic oscillators. The intensity of the wave is proportional to the square of the amplitude, so \begin{equation*} \text{Intensity}\propto e^{-2\omega n_Iz/c}. \end{equation*} This is often written as \begin{equation*} \text{Intensity}\propto e^{-\beta z}. \end{equation*} where $\beta=2\omega n_I/c$ is called the absorption coefficient. Thus we have in Eq. (32.33) not only the theory of the index of refraction of materials, but the theory of their absorption of light as well.
In what we usually consider to be transparent material, the quantity $c/\omega n_I$—which has the dimensions of a length—is quite large in comparison with the thickness of the material.
32–5The index of a mixture
There is another prediction of our theory of the index of refraction that we can check against experiment. Suppose we consider a mixture of two materials. The index of the mixture is not the average of the two indexes, but should be given in terms of the sum of the two polarizabilities, as in Eq. (32.34). If we ask about the index of, say, a sugar solution, the total polarizability is the sum of the polarizability of the water and that of the sugar. Each must, of course, be calculated using for $N$ the number per unit volume of the molecules of the particular kind. In other words, if a given solution has $N_1$ molecules of water, whose polarizability is $\alpha_1$, and $N_2$ molecules of sucrose (C$_{12}$H$_{22}$O$_{11}$), whose polarizability is $\alpha_2$, we should have that \begin{equation} \label{Eq:II:32:37} 3\biggl(\frac{n^2-1}{n^2+2}\biggr)= N_1\alpha_1+N_2\alpha_2. \end{equation}
We can use this formula to test our theory against experiment by measuring the index for various concentrations of sucrose in water. We are making several assumptions here, however. Our formula assumes that there is no chemical action when the sucrose is dissolved and that the disturbances to the individual atomic oscillators are not too different for various concentrations. So our result is certainly only approximate. Anyway, let’s see how good it is.
We have picked the example of a sugar solution because there is a good table of measurements of the index of refraction in the Handbook of Chemistry and Physics and also because sugar is a molecular crystal that goes into solution without ionizing or otherwise changing its chemical state.
| Data from Handbook | ||||||||
| A | B | C | D | E | F | G | H | J |
| Fraction of sucrose by weight | Density $(\text{g/cm}^3)$ | $n$ at $20^\circ$C | Moles of sucrosed per liter, $N_2/N_0$ | Moles of watere per liter, $N_1/N_0$ | $3\biggl(\dfrac{n^2-1}{n^2+2}\biggr)$ | $N_1\alpha_1$ | $N_2\alpha_2$ | $N_0\alpha_2$ |
| $0^\text{a}$ | $0.9982$ | $1.333\phantom{0}$ | $0$ | $55.5\phantom{0}$ | $0.617$ | $\underline{0.617}\phantom{0}$ | $0$ | — |
| $0.30$ | $1.1270$ | $1.3811$ | $0.989$ | $43.8\phantom{0}$ | $0.697$ | $0.488\phantom{0}$ | $0.209$ | $0.211$ |
| $0.50$ | $1.2296$ | $1.4200$ | $1.798$ | $34.16$ | $0.759$ | $0.380\phantom{0}$ | $0.379$ | $0.211$ |
| $0.85$ | $1.4454$ | $1.5033$ | $3.59\phantom{0}$ | $12.05$ | $0.887$ | $0.1340$ | $0.753$ | $0.210$ |
| $1.00^\text{b}$ | $1.588\phantom{0}$ | $1.5577^\text{c}$ | $4.64\phantom{0}$ | $0$ | $0.967$ | $0$ | $0.967$ | $0.208$ |
| apure water bsugar crystals caverage (see text) dmolecular weight of sucrose $=342$ emolecular weight of water $=18$ | ||||||||
We give in the first three columns of Table 32–2 the data from the handbook. Column A is the percent of sucrose by weight, column B is the measured density (g/cm$^3$), and column C is the measured index of refraction for light whose wavelength is $589.3$ millimicrons. For pure sugar we have taken the measured index of sugar crystals. The crystals are not isotropic, so the measured index is different along different directions. The handbook gives three values: \begin{equation*} n_1=1.5376,\quad n_2=1.5651,\quad n_3=1.5705. \end{equation*} We have taken the average.
Now we could try to compute $n$ for each concentration, but we don’t know what value to take for $\alpha_1$ or $\alpha_2$. Let’s test the theory this way: We will assume that the polarizability of water ($\alpha_1$) is the same at all concentrations and compute the polarizability of sucrose by using the experiment of values for $n$ and solving Eq. (32.37) for $\alpha_2$. If the theory is correct, we should get the same $\alpha_2$ for all concentrations.
First, we need to know $N_1$ and $N_2$: let’s express them in terms of Avogadro’s number, $N_0$. Let’s take one liter ($1000$ cm$^3$) for our unit of volume. Then $N_i/N_0$ is the weight per liter divided by the gram-molecular weight. And the weight per liter is the density (multiplied by $1000$ to get grams per liter) times the fractional weight of either the sucrose or the water. In this way, we get $N_2/N_0$ and $N_1/N_0$ as in columns D and E of the table.
In column F we have computed $3(n^2-1)/(n^2+2)$ from the experimental values of $n$ in column C. For pure water, $3(n^2-1)/(n^2+2)$ is $0.617$, which is equal to just $N_1\alpha_1$. We can then fill in the rest of Column G, since for each row G/E may be in the same ratio—namely, $0.617:55.5$. Subtracting column G from column F, we get the contribution $N_2\alpha_2$ of the sucrose, shown in column H. Dividing these entries by the values of $N_2/N_0$ in column D, we get the value of $N_0\alpha_2$ shown in column J.
From our theory we would expect all the values of $N_0\alpha_2$ to be the same. They are not exactly equal, but pretty close. We can conclude that our ideas are fairly correct. Even more, we find that the polarizability of the sugar molecule doesn’t seem to depend much on its surroundings—its polarizability is nearly the same in a dilute solution as it is in the crystal.
32–6Waves in metals
The theory we have worked out in this chapter for solid materials can also be applied to good conductors, like metals, with very little modification. In metals some of the electrons have no binding force holding them to any particular atom; it is these “free” electrons which are responsible for the conductivity. There are other electrons which are bound, and the theory above is directly applicable to them. Their influence, however, is usually swamped by the effects of the conduction electrons. We will consider now only the effects of the free electrons.
If there is no restoring force on an electron—but still some resistance to its motion—its equation of motion differs from Eq. (32.1) only because the term in $\omega_0^2x$ is lacking. So all we have to do is set $\omega_0^2=0$ in the rest of our derivations—except that there is one more difference. The reason that we had to distinguish between the average field and the local field in a dielectric is that in an insulator each of the dipoles is fixed in position, so that it has a definite relationship to the position of the others. But because the conduction electrons in a metal move around all over the place, the field on them on the average is just the average field $\FLPE$. So the correction we made to Eq. (32.8) by using Eq. (32.28) should not be made for conduction electrons. Therefore the formula for the index of refraction for metals should look like Eq. (32.27), except with $\omega_0$ set equal to zero, namely, \begin{equation} \label{Eq:II:32:38} n^2=1+\frac{Nq_e^2}{m\epsO}\,\frac{1}{-\omega^2+i\gamma\omega}. \end{equation} This is only the contribution from the conduction electrons, which we will assume is the major term for metals.
Now we even know how to find what value to use for $\gamma$, because it is related to the conductivity of the metal. In Chapter 43 of Volume I we discussed how the conductivity of a metal comes from the diffusion of the free electrons through the crystal. The electrons go on a jagged path from one scattering to the next, and between scatterings they move freely except for an acceleration due to any average electric field (as shown in Fig. 32–2). We found in Chapter 43 of Volume I that the average drift velocity is just the acceleration times the average time $\tau$ between collisions. The acceleration is $q_eE/m$, so \begin{equation} \label{Eq:II:32:39} v_{\text{drift}}=\frac{q_eE}{m}\,\tau. \end{equation} This formula assumed that $E$ was constant, so that $v_{\text{drift}}$ was a steady velocity. Since there is no average acceleration, the drag force is equal to the applied force. We have defined $\gamma$ by saying that $\gamma mv$ is the drag force [see Eq. (32.1)], which is $q_eE$; therefore we have that \begin{equation} \label{Eq:II:32:40} \gamma=\frac{1}{\tau}. \end{equation}
Although we cannot easily measure $\tau$ directly, we can determine it by measuring the conductivity of the metal. It is found experimentally that an electric field $\FLPE$ in a metal produces a current with the density $\FLPj$ proportional to $\FLPE$ (for isotropic materials): \begin{equation*} \FLPj=\sigma\FLPE. \end{equation*} The proportionality constant $\sigma$ is called the conductivity. This is just what we expect from Eq. (32.39) if we set \begin{equation} j=Nq_ev_{\text{drift}}.\notag \end{equation} Then \begin{equation} \label{Eq:II:32:41} \sigma=\frac{Nq_e^2}{m}\,\tau. \end{equation} So $\tau$—and therefore $\gamma$—can be related to the observed electrical conductivity. Using Eqs. (32.40) and (32.41), we can rewrite our formula for the index, Eq. (32.38), in the following form: \begin{equation} \label{Eq:II:32:42} n^2=1+\frac{\sigma/\epsO}{i\omega(1+i\omega\tau)}, \end{equation} where \begin{equation} \label{Eq:II:32:43} \tau=\frac{1}{\gamma}=\frac{m\sigma}{Nq_e^2}. \end{equation} This is a convenient formula for the index of refraction of metals.
32–7Low-frequency and high-frequency approximations; the skin depth and the plasma frequency
Our result, Eq. (32.42), for the index of refraction for metals predicts quite different characteristics for wave propagation at different frequencies. Let’s first see what happens at very low frequencies. If $\omega$ is small enough, we can approximate Eq. (32.42) by \begin{equation} \label{Eq:II:32:44} n^2=-i\,\frac{\sigma}{\epsO\omega}. \end{equation} Now, as you can check by taking the square,2 \begin{equation} \sqrt{-i}=\frac{1-i}{\sqrt{2}};\notag \end{equation} so for low frequencies, \begin{equation} \label{Eq:II:32:45} n=\sqrt{\sigma/2\epsO\omega}\,(1-i). \end{equation} The real and imaginary parts of $n$ have the same magnitude. With such a large imaginary part to $n$, the wave is rapidly attenuated in the metal. Referring to Eq. (32.36), the amplitude of a wave going in the z-direction decreases as \begin{equation} \label{Eq:II:32:46} \exp\bigl[-\sqrt{\sigma\omega/2\epsO c^2}\,z\bigr]. \end{equation} Let’s write this as \begin{equation} \label{Eq:II:32:47} e^{-z/\delta}, \end{equation} where $\delta$ is then the distance in which the wave amplitude decreases by the factor $e^{-1}=1/2.72$—or roughly one-third. The amplitude of such a wave as a function of $z$ is shown in Fig. 32–3. Since electromagnetic waves will penetrate into a metal only this distance, $\delta$ is called the skin depth. It is given by \begin{equation} \label{Eq:II:32:48} \delta=\sqrt{2\epsO c^2/\sigma\omega}. \end{equation}
Now what do we mean by “low” frequencies? Looking at Eq. (32.42), we see that it can be approximated by Eq. (32.44) only if $\omega\tau$ is much less than one and if $\omega\epsO/\sigma$ is also much less than one—that is, our low-frequency approximation applies when \begin{equation} \omega\ll\frac{1}{\tau}\notag \end{equation} and \begin{equation} \label{Eq:II:32:49} \omega\ll\frac{\sigma}{\epsO}. \end{equation}
Let’s see what frequencies these correspond to for a typical metal like copper. We compute $\tau$ by using Eq. (32.43), and $\sigma/\epsO$, by using the measured conductivity. We take the following data from a handbook: \begin{align*} &\sigma=5.76\times10^7\text{ (ohm$\cdot$meter)}^{-1},\\[2pt] &\text{atomic weight${}=63.5$ grams},\\[2pt] &\text{density${}=8.9$ grams$\cdot$cm$^{-3}$},\\[2pt] &\text{Avogadro’s number${}=6.02\times10^{23}$ (gram atomic weight)$^{-1}$}. \end{align*} \begin{align*} &\sigma=5.76\times10^7\text{ (ohm$\cdot$meter)}^{-1},\\[2pt] &\text{atomic weight${}=63.5$ grams},\\[2pt] &\text{density${}=8.9$ grams$\cdot$cm$^{-3}$},\\[2pt] &\text{Avogadro’s number${}=$}\\[2pt] &\phantom{Av}\text{$6.02\times 10^{23}$ (gram atomic weight)$^{-1}$}. \end{align*} If we assume that there is one free electron per atom, then the number of electrons per cubic meter is \begin{equation*} N=8.5\times10^{28}\text{ meter}^{-3}. \end{equation*} Using \begin{align*} q_e &=1.6\times10^{-19}\text{ coulomb},\\[2pt] \epsO &=8.85\times10^{-12}\text{ farad}\cdot\text{meter}^{-1},\\[2pt] m &=9.11\times10^{-31}\text{ kg}, \end{align*} we get \begin{align*} \tau &=2.4\times10^{-14}\text{ sec},\\[5pt] \frac{1}{\tau} &=4.1\times10^{13}\text{ sec}^{-1},\\[5pt] \frac{\sigma}{\epsO} &=6.5\times10^{18}\text{ sec}^{-1}. \end{align*} So for frequencies less than about $10^{12}$ cycles per second, copper will have the “low-frequency” behavior we describe (that means for waves whose free-space wavelength is longer than $0.3$ millimeters—very short radio waves!).
For these waves, the skin depth in copper is \begin{equation*} \delta=\sqrt{\frac{0.028\:\text{m}^2\cdot\text{sec}^{-1}}{\omega}} \end{equation*} For microwaves of $10{,}000$ megacycles per second ($3$-cm waves) \begin{equation*} \delta=6.7\times10^{-5}\text{ cm}. \end{equation*} The wave penetrates a very small distance.
We can see from this why in studying cavities (or waveguides) we needed to worry only about the fields inside the cavity, and not in the metal or outside the cavity. Also, we see why the losses in a cavity are reduced by a thin plating of silver or gold. The losses come from the current, which are appreciable only in a thin layer equal to the skin depth.
Suppose we look now at the index of a metal like copper at high frequencies. For very high frequencies $\omega\tau$ is much greater than one, and Eq. (32.42) is well approximated by \begin{equation} \label{Eq:II:32:50} n^2=1-\frac{\sigma}{\epsO\omega^2\tau}. \end{equation} For waves of high frequencies the index of a metal becomes real—and less than one! This is also evident from Eq. (32.38) if the dissipation term with $\gamma$ is neglected, as can be done for very large $\omega$. Equation (32.38) gives \begin{equation} \label{Eq:II:32:51} n^2=1-\frac{Nq_e^2}{m\epsO\omega^2}, \end{equation} which is, of course, the same as Eq. (32.50). We have seen before the quantity $Nq_e^2/m\epsO$, which we called the square of the plasma frequency (Section 7–3): \begin{equation*} \omega_p^2=\frac{Nq_e^2}{\epsO m}, \end{equation*} so we can write Eq. (32.50) or Eq. (32.51) as \begin{equation*} n^2=1-\biggl(\frac{\omega_p}{\omega}\biggr)^2. \end{equation*} The plasma frequency is a kind of “critical” frequency.
For $\omega<\omega_p$ the index of a metal has an imaginary part, and waves are attenuated; but for $\omega\gg\omega_p$ the index is real, and the metal becomes transparent. You know, of course, that metals are reasonably transparent to x-rays. But some metals are even transparent in the ultraviolet. In Table 32–3 we give for several metals the experimental observed wavelength at which they begin to become transparent. In the second column we give the calculated critical wavelength $\lambda_p=2\pi c/\omega_p$. Considering that the experimental wavelength is not too well defined, the fit of the theory is fairly good.
| Metal | $\lambda\text{(experimental)}$ | $\lambda_p=2\pi c/\omega_p$ |
| $\text{Li}\;$ | $1550$ Å | $1550$ Å |
| $\text{Na}$ | $2100\phantom{00}$ | $2090\phantom{00}$ |
| $\text{K}\phantom{b}$ | $3150\phantom{00}$ | $2870\phantom{00}$ |
| $\text{Rb}$ | $3400\phantom{00}$ | $3220\phantom{00}$ |
| *From: C. Kittel, Introduction to Solid State Physics, John Wiley and Sons, Inc., New York, 2nd ed., 1956, p. 266. | ||
You may wonder why the plasma frequency $\omega_p$ should have anything to do with the propagation of electromagnetic waves in metals. The plasma frequency came up in Chapter 7 as the natural frequency of density oscillations of the free electrons. (A clump of electrons is repelled by electric forces, and the inertia of the electrons leads to an oscillation of density.) So longitudinal plasma waves are resonant at $\omega_p$. But we are now talking about transverse electromagnetic waves, and we have found that transverse waves are absorbed for frequencies below $\omega_p$. (It’s an interesting and not accidental coincidence.)
Although we have been talking about wave propagation in metals, you appreciate by this time the universality of the phenomena of physics—that it doesn’t make any difference whether the free electrons are in a metal or whether they are in the plasma of the ionosphere of the earth, or in the atmosphere of a star. To understand radio propagation in the ionosphere, we can use the same expressions—using, of course, the proper values for $N$ and $\tau$. We can see now why long radio waves are absorbed or reflected by the ionosphere, whereas short waves go right through. (Short waves must be used for communication with satellites.)
We have talked about the high- and low-frequency extremes for wave propagation in metals. For the in-between frequencies the full-blown formula of Eq. (32.42) must be used. In general, the index will have real and imaginary parts; the wave is attenuated as it propagates into the metal. For very thin layers, metals are somewhat transparent even at optical frequencies. As an example, special goggles for people who work around high-temperature furnaces are made by evaporating a thin layer of gold on glass. The visible light is transmitted fairly well—with a strong green tinge—but the infrared is strongly absorbed.
Finally, it cannot have escaped the reader that many of these formulas resemble in some ways those for the dielectric constant $\kappa$ discussed in Chapter 10. The dielectric constant $\kappa$ measures the response of the material to a constant field, that is, for $\omega=0$. If you look carefully at the definition of $n$ and $\kappa$ you see that $\kappa$ is simply the limit of $n^2$ as $\omega\to0$. Indeed, placing $\omega=0$ and $n^2=\kappa$ in equations of this chapter will reproduce the equations of the theory of the dielectric constant of Chapter 11.
- Throughout this chapter we follow the notation of Chapter 31 of Volume I, and let $\alpha$ represent the atomic polarizability as defined here. In the last chapter, we used $\alpha$ to represent the volume polarizability—the ratio of $P$ to $E$. In the notation of this chapter $P=N\alpha\epsO E$ (see Eq. 32.8). ↩
- Or writing $-i=e^{-i\pi/2}$; $\sqrt{-i}=$ $e^{-i\pi/4}=$ $\cos\pi/4-i\sin\pi/4$, which gives the same result. ↩